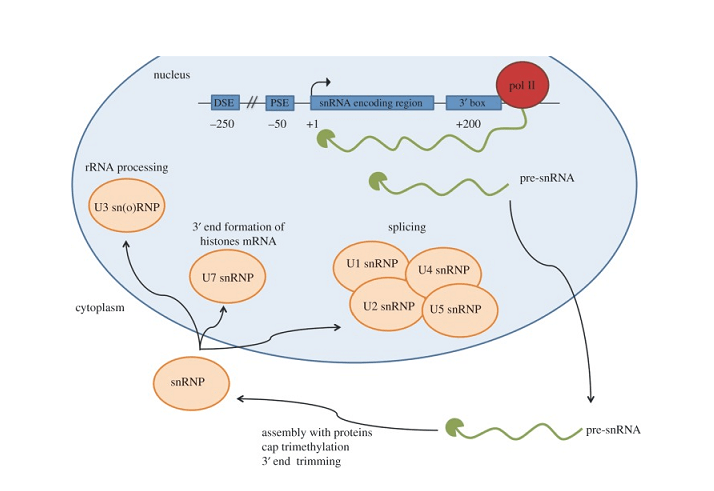
Single nuclear RNAs, or snRNAs, play a crucial role within cells despite their short lengths. As non-coding RNA molecules found solely in the nucleus, snRNAs participate in essential post-transcriptional modifications that help synthesize functional proteins. There are two main classes of snRNAs – U1, U2, U4, U5, and U6 which form the small nuclear ribonucleoproteins (snRNPs) involved in splicing, and the H/ACA and C/D box snoRNAs which guide chemical modifications of other RNAs.
What is single-nucleus RNA sequencing (snRNA-seq)?
Single-nucleus RNA sequencing, or snRNA-seq, is an innovative technique that analyzes the nuclear transcriptome isolated from individual cells. This approach helps minimize transcriptional stress responses that can skew results during dissociation. Compared to traditional single-cell RNA sequencing (scRNA-seq), snRNA-seq exhibits certain benefits that enhance its utility for investigating cellular heterogeneity.
What is the Difference Between snRNA-seq and scRNA-seq?
Single-nucleus RNA sequencing (snRNA-seq), also known as snRNA-seq or sNuc-seq, is an innovative RNA sequencing method used to profile gene expression in cells that are challenging to isolate. This technique serves as a powerful alternative to single-cell RNA sequencing (scRNA-seq) by analyzing nuclei instead of intact cells. But what exactly sets snRNA-seq apart from scRNA-seq? Let’s delve into the key differences.
scRNA-seq measures both cytoplasmic and nuclear transcripts, providing a comprehensive view of a cell’s gene expression profile. In contrast, snRNA-seq primarily measures nuclear transcripts, as it focuses on analyzing the RNA within the nucleus itself. This targeted approach minimizes the occurrence of spurious gene expression, as mature ribosomes reside in the cytoplasm. Por lo tanto, any transcription factors expressed after the dissociation process cannot be translated, preventing the transcription of their downstream targets.
How does snRNA-seq accommodate challenging tissue types?
For one, snRNA-seq accommodates a wider variety of tissue types that may be challenging to completely dissociate into single cells. Complex tissues with dense extracellular matrices like the brain, kidney, and heart can be analyzed more effectively using snRNA-seq due to its milder dissociation protocol. This preserves the integrity of sensitive cell types that may undergo transcriptional changes or degrade during harsher dissociation required for scRNA-seq.
What are the benefits of using snRNA-seq for archived samples?
SnRNA-seq also supports the study of archived clinical samples. Frozen or fixed tissues that would be incompatible with scRNA-seq can be profiled as long as intact nuclei can be isolated. This expands the scope of possible research applications compared to other sequencing methods that demand freshly isolated single cells. Notably, snRNA-seq has enabled investigators to analyze post-mortem human brain tissue to uncover neuronal diversity on a single-cell level.
What unique perspective does snRNA-seq provide?
Rather than a comprehensive view of the full transcriptome, snRNA-seq provides focused insights into nuclear processes and interactions modulated by snRNAs. It offers a uniquely magnified perspective on regulatory mechanisms within the confined nuclear space. Applying snRNA-seq to complex tissue samples has revealed new cell types and states that may go undetected using scRNA-seq alone. Such discoveries deepen our understanding of cellular organization and specialization, expanding the frontiers of precision medicine.
How can snRNA-seq help profile gene expression in challenging samples?
The nuclear profiling capabilities of snRNA-seq prove especially valuable for samples that are problematic to analyze using conventional scRNA-seq approaches. Certain tissue types pose technical difficulties due to qualities like dense matrix composition, fragility upon dissociation, or incompatibility with enzymatic treatments required for single-cell isolation. archived samples preserved through fixation also present challenges.
How was snRNA-seq used to study mammalian heart tissue?
Mammalian heart tissue exemplifies a system intractable to scRNA-seq yet amenable to snRNA-seq analysis. Cardiac muscle cells form an intricate interconnected network not easily disrupted without cellular damage. Sin embargo, en 2020 German researchers successfully sequenced the first adult mammalian heart using snRNA-seq. By isolating nuclei instead of whole cardiomyocytes, they obtained practical distributions of distinctive cell populations within the myocardium. Such profiling at scale would be impractical through scRNA-seq methodology.
How does snRNA-seq benefit the study of neuronal populations?
Neuronal populations represent another class benefiting from snRNA-seq capabilities. The brain consists of intricately wired neurons not readily dissociated as intact single cells. Attempting to do so can induce stress responses skewing results. Yet studying the transcriptomic heterogeneity of neural subtypes informs our understanding of neurobiology, disease mechanisms, y más. SnRNA-seq protocols optimized for CNS tissue preserve both archived and freshly resected human brain samples on a single-cell resolution.
How does snRNA-seq enable the analysis of frozen clinical samples?
Frozen clinical samples similarly pose limitations overcome by snRNA-seq. Liver disease studies comparing healthy and fibrotic lung nuclei detected via snRNA-seq revealed unbiased identification of constituent cell types. Such analyses of archived patient biospecimens were previously excluded from analysis due to scRNA-seq input constraints. Now researchers can apply ‘omics insights to a wider range of real-world samples influencing precision diagnostics and therapy.
How does the snRNA-seq protocol minimize transcriptional perturbations?
A major advantage of snRNA-seq lies in its gentler dissociation protocols that forestall technical problems arising from heat, prolonged incubation times or excessive enzymatic treatments. The brief, mild lysis method employed for nuclear separation prevents stress-induced transcriptional changes that can skew scRNA-seq results. Without subjecting intact cells to harsh dissociation steps, snRNA-seq sampling better preserves the integrity of transcripts present at the time of dissociation.
How does snRNA-seq minimize spurious gene expression patterns?
This minimizes the occurrence of spurious gene expression patterns caused by the dissociation process itself. Mature ribosomes found solely within cytoplasmic compartments means any mRNAs encoding immediate-early stress response genes expressed post-dissociation cannot be translated. Their downstream transcriptional targets thus remain unaltered. Comparative studies applying both techniques to the same tissue have shown that snRNA-seq captures a wider subset of cell types with less dissociation-induced transcriptional perturbation.
How do short lysis durations help control transcriptomic biases in snRNA-seq?
Additional factors like short lysis durations help control transcriptomic biases. Prolonged exposure on ice before proceeding with single-nucleus library preparation can negatively impact data quality by initiating transcriptional changes. Careful timing assuages such effects. Sufficient initial tissue amounts also assure high-quality nuclei preparations necessary for robust profiling given methodological limitations.
How do optimized snRNA-seq workflows minimize technical artifacts?
Overall, optimized snRNA-seq workflows minimize pre-analytical factors complicating biological interpretation. Their gentle extraction of nuclear content presents gene expression profiles shaped less by technical artifacts than endogenous biological processes. This lends greater clarity and confidence to discoveries gleaned regarding regulatory networks, rare subpopulations, and cellular specialization programs within complex in vivo systems.
How can researchers apply snRNA-seq to advance biological understanding?
As an innovative technique refining our view of cellular complexity, snRNA-seq opens new frontiers for diverse research applications. A few examples highlight its potential for driving scientific progress:
- Developmental biology: Comparing nuclei from distinct embryonic stages could reveal stage-specific regulatory programs guiding differentiation. Spatially resolved snRNA-seq mapping developmental gradients may uncover patterning mechanisms.
- Disease pathology: Profiling diseased versus healthy tissue nuclei may identify pathology-associated changes in rare cell types illuminating disease mechanisms. Patient sample analyses could inform precision diagnostics and therapeutics.
- Neuropsychiatry: Investigating archived post-mortem brain tissue via snRNA-seq helps characterize the neurodiversity of psychiatric conditions. Longitudinal studies may uncover longitudinal biomarkers and therapeutic targets.
- Evolutionary studies: SnRNA-seq makes feasible cross-species comparisons previously challenging due to inter-organism differences in tissue properties. Such “phylo-snRNA-seq” informs our understanding of conserved versus divergent regulatory processes.
- Multi-omics integration: Pairing snRNA-seq with spatial profiling techniques yields spatially resolved epigenomic and transcriptomic maps elucidating nuclear microenvironments. Integrating spatial transcript data with proteomic or metabolomic layers provides systems-level context.
Resumen
As one of several emerging techniques revolutionizing single-cell analysis, snRNA-seq catalyzes discoveries by making previously intractable samples experimentally tractable. Its scaling up of nuclear omics promises fresh insights advancing diverse fields from regenerative medicine to precision oncology. Together with complementary approaches, snRNA-seq helps unravel life’s fascinating complexity on an unprecedented level.