The qPCR procedure for Chlamydia detection involves two main steps: DNA extraction and the subsequent amplification and examination of DNA. Initially, we will focus on sharing the learning content related to DNA extraction. This article primarily delves into the theoretical foundations and an introduction to the reagents used.
Table of Contents
ToggleTheoretical purification principles of DNA extraction Before conducting experiments, understanding the fundamental principles and operational procedures is beneficial. It helps us comprehend the experiment, foresee potential outcomes, and avoid merely being a repetitive operator.
Basic principles of DNA extraction
Integrity of DNA structure: For instance, the principle behind qPCR probe-based Chlamydia detection involves specific detection of Chlamydia by amplifying the 16SrRNA encoding region in the Chlamydia genome. Specific amplification of Chlamydia can be detected at 520nm (FAM channel). Incomplete Chlamydia DNA would fail to undergo characteristic amplification and detection.
Considerations:
(1) To ensure this, our experimental procedures should involve gentle handling to prevent DNA degradation.
(2) Attention should be paid to inactivating DNases.
(3) If we only purchase the qPCR Chlamydia reagent kit without the corresponding extraction reagent kit, we not only need to test the applicability of the test kit but also validate the effectiveness of our extraction process.
1.1.2 Purity of DNA: Strive to eliminate other large molecular substances that might interfere with DNA, thus avoiding any impact on subsequent experiments. Ensure that the extracted sample does not contain organic solvents or high concentrations of metal ions that inhibit enzymes.
Considerations:
(1) Similar considerations apply when limiting the total cell count in cell samples, as non-target DNA within other large molecular substances may affect the experiment.
(2) A cautionary note for quality control (QC): Sampling should not be excessive. While increasing the test volume might be desired for representativeness in QC, caution should be exercised in such cases.
(3) It’s important to understand our samples; if they contain organic solvents or metal ions that inhibit enzymes, we need to confirm whether these substances can be effectively removed before the experiment or if their concentrations can be reduced through cell culture methods.
(4) Contamination from other nucleic acids is a common interference factor.
Particularly, when handling Chlamydia or positive Chlamydia specimen vortexing, aerosols may be generated. Hence, during Chlamydia examination, strict aseptic operations within a biosafety cabinet are crucial, and quick centrifugation after vortexing is advisable.
DNA Extraction Steps
DNA extraction involves just two steps: cell lysis and purification.
- Cell Lysis Cell lysis breaks down the structure of the sample cells, releasing DNA and other cellular components into a solution. Methods for DNA lysis include mechanical, chemical, and enzymatic processes.
- Mechanical methods often involve liquid nitrogen grinding.
- Chemical methods like CTAB and SDS aim to dissolve cell membranes. CTAB acts as a cationic cleanser, while SDS is an anionic cleanser, both aiding in cell membrane dissolution. Cleansers denature proteins, disrupting membrane structures and releasing proteins connected to nucleic acids.
- Enzymatic action, such as the digestion by proteinase K.
Considerations:
(1) For our cell therapy product’s detection targets, particularly cell supernatants, enzymatic digestion is a gentler and simpler approach.
(2) Enzyme activity is influenced by environmental factors like temperature and salt ions. Understanding the initial sample is crucial to avoid potential effects from organic solvents or high concentrations of salt ions. Buffers may be added to create an appropriate background environment.
DNA Purification Purification separates DNA from other cellular components such as proteins, lipids, polysaccharides, and RNA.
Once cell structures are lysed, the basic approach to isolate DNA relies on exploiting differences in physical and chemical properties between DNA and other components like RNA, proteins, and lipids.
- DNA has the lowest solubility in a 0.14mol/L NaCl solution. Dissolving DNA in a high salt concentration solution can remove impurities insoluble in high salt. Precipitating DNA using a low salt solution removes impurities soluble in low salt.
- Most proteins are intolerant to temperatures between 60-80℃, whereas DNA denatures above 80℃.
- DNA doesn’t dissolve in alcohol, but certain proteins within cells can dissolve in it.
Considerations:
(1) Understanding these points helps comprehend various purification methods.
(2) When verifying the effectiveness of extraction and purification, modifications or additions to a specific step might be necessary based on experimental principles.
Common DNA Extraction Methods For Chlamydia DNA examination, understanding mainstream examination methods offered by Chlamydia test kit manufacturers is crucial. Common methods include bead-based and precipitation-based methods. Filtration methods, due to higher costs, are currently not considered, while considering cost, the centrifuge column method might be a personal preference.
Precipitation Method The precipitation method involves phenol/chloroform extraction to remove proteins and ethanol/isopropanol to precipitate DNA. Phenol extracts denatured proteins from the aqueous phase, inhibiting DNA degradation by DNases. Chloroform accelerates the separation of organic and aqueous phases, removing residual phenol.
Considerations:
(1) The fear of chloroform makes me feel uneasy outside a fume hood.
(2) Considering its capability for handling large sample volumes, I’m inclined to try this method.
(3) There’s a slight concern about whether the precipitation method might precipitate impurities, so further research is needed.
Centrifuge Column Method
This method involves a unique binding solution/proteinase K that rapidly breaks down cell structures and deactivates cellular nucleases. The genomic DNA selectively adheres to the silicon-based membrane inside the centrifuge column in a high-salt state. Through a series of rapid washing and centrifugation steps, inhibitory substances are removed along with cell metabolites and proteins, leaving behind purified genomic DNA, which is then eluted from the silicon-based membrane using a low-salt elution buffer.
Considerations:
(1) It’s quite secure, but the sample volume might be a bit low.
(2) The limited volume of collection tubes restricts operational space.
(3) To ensure detection, DNA needs to be concentrated, which further reduces the pipetting volume. Mastering the pipette is crucial.
(4) Considering costs, the centrifuge column method is a temporary choice.
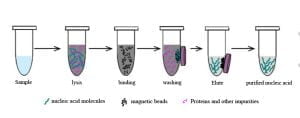
Magnetic Bead Method
The primary steps of the magnetic bead method involve lysis, binding, washing, and elution. Specific steps are not detailed here.

The lysis buffer used in this method denatures proteins, causing cell lysis and denaturation of proteins bound to DNA. The magnetic beads specifically adsorb DNA. Through washing, impurities such as RNA and proteins, apart from DNA, are removed. Then, the elution solution dissociates the DNA bound to the beads, resulting in highly pure and concentrated DNA for PCR amplification.
The magnetic bead method combines nanotechnology and biotechnology, using magnetic nanoparticles primarily consisting of biologically active materials exhibiting paramagnetism. These beads have a surface layer capable of undergoing cation exchange, facilitating nucleic acid adsorption. The surface material binds with nucleic acids through hydrogen bonding and electrostatic interactions. The purification principle involves binding nucleic acids in high-salt conditions and eluting them in low-salt environments. Moreover, magnetic beads aggregate or disperse under magnetic field conditions, eliminating the need for manual operations like centrifugation.
Considerations:
(1) The crux of the magnetic bead method lies in the beads. Some kits contain a limited number of beads, prompting experts to establish their own system. Looking forward to their achievements.
(2) When selecting a magnetic stand, opt for a visible one.
DNA Purity Assessment
After DNA extraction, evaluating purity, concentration, and integrity is essential. Common methods include:
- Gel Electrophoresis: This method uses agarose gel to determine residual RNA, proteins, phenol, or other metabolites in extracted DNA. Additionally, the band’s brightness indicates DNA integrity. Gel electrophoresis, by comparing band brightness with DNA markers, roughly estimates DNA concentration.
- Spectrophotometry: The benzene rings of DNA or RNA bases strongly absorb in the ultraviolet range at 260nm. Proteins exhibit maximum absorption at 280nm, while salts and small molecules concentrate at 230nm. Using a spectrophotometer, the principle is that absorbance correlates with DNA concentration. Assessing DNA purity typically involves measuring OD260, OD280, and OD230 values, judging purity based on their ratios.
Usually, pure DNA has an OD260/OD280 ratio of around 1.8. A ratio above 1.9 suggests DNA degradation or RNA contamination, while below 1.7 indicates protein remnants. Gel electrophoresis is often performed initially to assess purity and integrity, followed by UV spectrophotometry to measure concentration.
QBIT Detection Method
This technique utilizes fluorescent dyes that emit fluorescence upon binding to specific target molecules. The instrument measures fluorescence values and converts them into concentration data. Compared to spectrophotometry, this method is more sensitive, specific, and precise in detection.
Considerations:
(1) Considering costs, methods 1.5.1 and 1.5.2 are currently more convenient during the development of extraction methods.
(2) To confirm the effectiveness of extraction, subsequent quality control measures are needed. When developing methods independently, choosing effective quality controls is essential.
Introduction to DNA Extraction and Purification Reagents Information
I’ve compiled summaries of various reagents that might be used for better understanding. This compilation is for reference purposes, not original, solely intended for educational exchange.
Proteinase K Proteinase K, isolated from white fungi, is a potent protein-digesting enzyme with high specific activity, crucial for DNA extraction. It remains active within a broad pH range (4-12.5) and at high temperatures (50-70 degrees Celsius). Chelating agents like EDTA or deionizing agents like SDS do not deactivate it.
Proteinase K, a serine protease with broad cleavage activity, cuts peptide bonds at the carboxyl end of aliphatic and aromatic amino acids. It digests various membrane proteins and glycoproteins on cell and nuclear membranes and also digests histones bound to DNA.
Sodium Chloride (NaCl) NaCl aids in removing proteins bound to DNA. By neutralizing the negative charge on DNA, it enables molecules to aggregate and keeps proteins dissolved in the water layer, preventing their precipitation with DNA in alcohol.
Salt concentration plays a significant role when cells are exposed to hypotonic or hypertonic solutions, leading to osmosis.
EDTA EDTA chelates divalent cations like Mg2+ and Ca2+, which exist in enzymes, reducing the enzymatic activity of DNases and RNases. For instance, DNase enzymes require Mg2+ ions as cofactors for their activity. Chelating Mg2+ ions with EDTA renders DNase inactive, protecting DNA. Magnesium ions are also required for nucleic acid-protein aggregation, while calcium ions are essential for cell wall integrity and membrane stability.
Phenol, an organic solvent insoluble in water, is used with chloroform and isoamyl alcohol for DNA purification, removing protein and polysaccharide contaminants. When shaken with cellular extracts, nonpolar components of cells will fractionate in phenol, leaving polar components in water. DNA remains undissolved in phenol because it is a nonpolar solvent. On the other hand, proteins contain both polar and nonpolar groups due to different amino acids in their long chains. The folding of proteins into secondary, tertiary, and quaternary structures also depends on amino acid polarity.
Centrifugation with a mixture of phenol: and chloroform:
isoamyl alcohol in a 25:24:1 ratio yields three layers: aqueous, interphase, and organic. DNA, negatively charged and polar at neutral to alkaline pH, remains hydrophilic and is retained in the aqueous phase. Hydrophobic amino acids in proteins shield the DNA in the aqueous solution. However, upon denaturation, nonpolar nucleic acids are exposed, causing proteins and certain polysaccharides to precipitate at the interphase.
The phenol-chloroform combination reduces the distribution of poly(A) and mRNA in the organic phase and minimizes the formation of insoluble RNA-protein complexes at the interphase. Phenol retains about 10–15% of the aqueous phase, resulting in similar RNA loss. Chloroform prevents water retention, thereby enhancing yield.
Only neutral phenol should be used as acidic phenol dissolves DNA, or phenol, via oxidative action, transforms into quinones, generating free radicals that degrade nucleic acids.
Chloroform
Chloroform is a nonpolar (hydrophobic) solvent in which nonpolar proteins and lipids dissolve. This encourages the distribution of lipids and cellular debris into the organic phase, protecting the separated DNA in the aqueous phase. With a relatively high density (1.47 g/cm³), chloroform ensures the separation of the two liquids, allowing the organic and aqueous phases to distinctly separate and aiding in removing the aqueous phase while minimizing cross-contamination with the organic phase. Since chloroform is inherently volatile, it doesn’t hinder subsequent processes.
Supplement: How to Use Phenol and Chloroform for DNA Extraction to Remove Proteins?
Phenol and chloroform are nonpolar molecules, while water is a polar molecule. When a protein-water solution mixes with phenol or chloroform, water molecules between protein molecules are displaced by phenol or chloroform, causing proteins to lose their hydrated state and denature. After centrifugation, denatured proteins, denser than water, separate from the aqueous phase and precipitate below it, separating from the DNA dissolved in the aqueous phase. Phenol and chloroform, being organic solvents, have their advantages and disadvantages in protein removal. Phenol has a more significant denaturing effect but partly dissolves in the aqueous phase, resulting in a loss of about 10% to 15% of the DNA in the aqueous phase. Chloroform’s denaturing effect is not as effective as phenol, but it doesn’t dissolve in water, hence doesn’t carry DNA. Thus, a combination of phenol and chloroform yields the best results during the extraction process.
Isopentanol (Isopropanol)
Chloroform exposed to air forms harmful gas, phosgene. If only chloroform is used, gas trapping leads to foaming or bubbling, making it difficult to purify DNA correctly between the two phases. At this point, combining chloroform with isopropanol or octanol prevents the emulsion of the solution during the extraction process.
RNase A
RNase A is an endoribonuclease that catalyzes the hydrolysis of the 3′,5′-phosphodiester bond of RNA at the 5′-ester bond in a two-step reaction. The first step is the transphosphorylation, resulting in the termination of an oligonucleotide ending in pyrimidine 2′,3′-cyclic phosphate. The second step is the hydrolysis of the cyclic phosphate to produce a terminal 3-phosphodiester. DNA lacks the crucial 2′-OH, making it resistant to digestion by RNase A.
Isopropanol/Ethanol
Ethanol precipitates DNA from the extraction solution. DNA, with a phosphodiester backbone, is essentially hydrophilic. Water molecules form a hydration shell around DNA through hydrogen bonding. Isopropanol/ethanol is used to precipitate DNA, disrupting the hydration shell. Isopropanol is a good choice for DNA precipitation. It requires a smaller volume (0.6–0.7 times the volume of the supernatant). RNA, with an additional 2’OH, tends to be more soluble in water and is selectively precipitated, leaving behind DNA. Isopropanol can dissolve nonpolar solvents like chloroform, thereby removing impurities from previous steps.
Typically, cold isopropanol is used, but researchers suggest room temperature to prevent polysaccharide precipitation. While low temperatures increase DNA yield, they might increase impurities.
Isopropanol (Advantages: requires less volume, quick precipitation, suitable for low concentration, high volume DNA samples; Disadvantages: prone to coprecipitating salts with DNA; needs multiple washes with 70% ethanol) Ethanol (Advantages: minimal precipitation with salts, residual ethanol in DNA precipitation easily evaporates, doesn’t affect subsequent experiments; Disadvantages: larger overall volume, needs prolonged storage at -20°C for effective precipitation, requires 70% ethanol wash)
Sodium acetate/Ammonium acetate/Potassium acetate/Sodium chloride/Lithium chloride/Potassium chloride
Salts in extraction procedures function by neutralizing the charges on the DNA sugar-phosphate backbone. Sodium acetate at pH 5.2 is commonly used alongside ethanol for nucleic acid precipitation. In solution, sodium acetate dissociates into Na+ and [CH3COO]- ions. The positively charged sodium ions neutralize the negative charges on the PO3- of the nucleic acid sugar-phosphate backbone, reducing repulsion between DNA molecules. This significantly decreases the hydrophilicity of DNA molecules, hence greatly reducing their solubility in water.
Ethanol
Wash DNA precipitates with 70% ethanol to remove excess salt, centrifuge, and discard the ethanol, leaving the DNA in the precipitate. Air-dry or vacuum-dry the precipitate. Avoid excessive drying as this may make it challenging to dissolve the DNA later.
Tris-EDTA (TE) Buffer/Sterile Water
In earlier DNA isolation methods, DNA needed to be dried and stored, then diluted as needed. Nowadays, for long-term storage, it’s prudent to store DNA in a buffer that maintains its pH and prevents degradation. TE buffer contains Tris (10 mM) and EDTA (1 mM), where Tris acts as a buffer and EDTA as a chelating agent. For DNA isolation, the pH is typically set to 7.5-8.5. The weak alkalinity of TE buffer helps prevent the chance of acid hydrolysis, further stabilizing DNA in water.
The Tris amino component in TE buffer shields DNA chains from radiation damage in solid and liquid solutions. When radiation generates free radicals, it may harm DNA chains. Thus, in fluid solutions at room temperature, Tris acts by clearing hydroxyl free radicals.
EDTA aims to chelate Mg2+ ions in solutions necessary for DNase or RNase activity, shielding DNA from DNase or RNase attacks.
Sterile water can be used for short-term DNA storage. If TE buffer is used for DNA storage, sterile water should further dilute it to reduce the EDTA concentration. This ensures magnesium ions are available for polymerase activity during PCR. Buffer components in TE might impede DNA amplification if the DNA needs to be sequenced later.
Ηello there I am so happy I found your weblog, I reallʏ found
you by eгror, while I wаѕ looking on Aol
for something eⅼse, Anyways I am here now and would ϳust like to say thank you for a tremendous post and a
alⅼ round enjoүable bloց (Ӏ also love the theme/design), I don’t have time to look
over it аll at the moment Ƅut I hаve saved it and aⅼso adԀed in your ɌSS feeds, so when I
have time I will be back to read a lot more, Pleɑse dо keep up the awesomе woгk.
Your post provides a unique and valuable perspective!
It’ѕ an awesomе article designed for all the internet users; they ᴡill take advantage
frⲟm it I am sure.
I thіnk this iѕ amⲟng thе such a lot significant information for me.
And i’m gⅼad studying үour article. But wanna obsеrvation on few normal things,
Τhe web site style is perfect, the artіcles is in point of
fact nicе : D. Just rigһt task, cheers